Olga A. Omelchuk,a,b@1 Lyudmila N. Lysenkova,a Nikita M. Belov,а Alexander M. Korolev,a Lyubov G. Dezhenkova,a Natalia E. Grammatikova,a,c Olga B. Bekker,d Valery N. Danilenko,d and Andrey E. Shchekotikhina,b@2
aGause Institute of New Antibiotics, 119021 Moscow, Russian Federation
bMendeleev University of Chemical Technology of Russia, 125047 Moscow, Russian Federation
cSechenov First Moscow State Medical University, 119991 Moscow, Russian Federation
dVavilov Institute of General Genetics, Russian Academy of Sciences, 119333 Moscow, Russian Federation
@1Corresponding author E-mail: omelchuk.93@mail.ru
@2Corresponding author E-mail: shchekotikhin@mail.ru
DOI: 10.6060/mhc180795o
Macroheterocycles 2018 11(3) 322-328
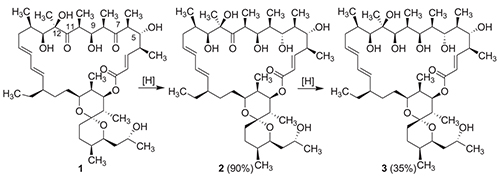
Macrolide antibiotics represent a valuable class of broad-spectrum, high active natural compounds with polyketide structure. A well-known FOF1 ATP-synthase inhibitor,[1] namely oligomycin A (1), is a 26-membered α,β-unsaturated polyketide lactone with conjugated diene, fused to spiroketal moiety. Oligomycin A possesses strong antifungal, antiactinomycotic and cytotoxic activity, but lacks antibacterial activity. According to recent investigations, the development of anti-cancer drugs based on oligomycin A is quite perspective due to its high cytotoxic activity toward tumor cells, ability to inhibit a multidrug resistance protein p-gp and to prevent an activation of oncogenic K-Ras by inhibition of its localization at the plasma membrane.[2-4] However, high toxicity for mammalian cells and low water solubility are significant limitations of oligomycin A, making it unacceptable for clinical application. Chemical modification is a promising way to improve pharmacological properties of natural compounds. Recently we have found that site-selective modifications of oligomycin A afforded semi-synthetic derivatives with high antiproliferative activity against tumor cell lines[5-7] or selective antifungal activity against Candida spp.[8] and, at the same time, with lower toxicity toward mammalian cells. Also, semi-synthetic oligomycin A derivatives are useful tools for molecular genetic studies of additional targets for this family of antibiotics.[9,10] Previously Ramirez F. et al. have described the reaction of oligomycins with sodium borohydride resulting in mixture of diastereomeric 7-dihydro- and 7,11-tetrahydro derivatives without further separation and characterization of individual products.[11] Also, there is no data on biological activity of these reduced oligomycins against fungal/actinomycetes strains and tumor cell lines in article mentioned above. Thus, in this paper we report regio- and stereoselective methods for borohydride reduction of oligomycin A, structure determination of obtained derivatives and investigation of theirs antiproliferative, antifungal and antiactinomycotic properties. The feasibility of regio- and stereoselective reduction of C7-carbonyl group in a core structure of oligomycin A was proposed due to the presence of haptophilic hydroxyl groups[12] at C5 and C9 positions and sterical hindrance of C-11 carbonyl group. Actually, treatment of oligomycin A with bulky sodium triacetoxyborohydride in acetic acid according to the method[13] led to (7S)-dihydrooligomycin A (2) in a good yield. The second carbonyl group (C-11) reduced in more harsh conditions: only the extended treatment of (7S)-dihydrooligomycin A with sodium borohydride in ethanol give (7S,11R)-7,11-tetrahydrooligomycin A (3) as major product. Reaction proceeds with acceptable stereoselectivity and gives tetrahydro derivative 3, but in low yield (35 %), which associated with low stability of oligomycins in basic conditions.[14] Structure of compounds 2 and 3 was confirmed by high resolution mass spectrometry (HRMS ESI) and NMR spectroscopy. Absolute configurations at C7 and C11 positions of obtained derivatives were unambiguously confirmed by observed interactions between neighboring protons in corresponding 1H-1H ROESY spectra. Testing of antimicrobial properties of oligomycins 2 and 3 against Candida spp., filamentous fungi and S. fradiae (strain, extremely sensitive to oligomycins) that of the parent antibiotic in comparison with starting oligomycin A revealed that reduction of carbonyl groups led to decreasing of activity (except strain M. canis). Also, reduced derivatives 2, 3 were less potent against human colon carcinoma cell line HCT116 and its doxorubicin-resistant subline HCT116(-/-), while activity against leukemia cell line K562 and doxorubicin-resistant subline K562/4 retained at the same level as for 1. It might be pointed that biological properties of (7S)-dihydrooligomycin A and (7S,11R)-7,11-tetrahydrooligomycin A are quite similar, consequently C7-carbonyl group has a greater influence on biological activity of oligomycin A than C-11 carbonyl group.
References: